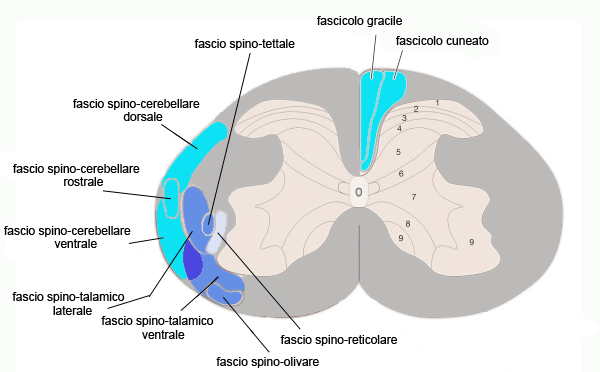
I fasci ascendenti del midollo spinale
I fasci (tratti) ascendenti (sensitivi) del midollo spinale sono situati nella sostanza bianca del cordone (funicolo) dorsale e laterale.
Il cordone dorsale contiene pressoché esclusivamente fasci ascendenti, eccezion fatta per i fascicoli setto-marginale e semilunare. I fasci ascendenti del cordone dorsale sono due: il fascicolo gracile e cuneato costituti dai prolungamenti centripeti dei neuroni sensitivi di primo ordine dei gangli spinali che danno origine alla via della colonna dorsale-lemnisco mediale. Si tratta di neuroni di grosse dimensioni i cui prolungamenti periferici formano recettori corpuscolati a livello cutaneo, fusi neuromuscolari, organi tendinei di Golgi e terminazioni articolari a livello dell’apparato locomotore.
Il cordone laterale contiene diversi fasci ascendenti che si localizzano più superficialmente rispetto ai fasci discendenti. I più importanti sono i fasci spino-cerebellari, i fasci spino-talamici, il fascio spino-reticolare, il fascio spino-olivare e il fascio spino-tettale. Tutti questi fasci derivano da neuroni sensitivi di secondo ordine situati in parti diverse della sostanza grigia del midollo spinale. Questi neuroni ricevono afferenze da neuroni sensitivi di primo ordine di grosse dimensioni situati nei gangli spinali. Tali neuroni funzionalmente agiscono come meccanocettori.
I fasci spino-cerebellari veicolano stimoli propriocettivi inconsci provenienti dalla muscolatura somatica e dalle articolazioni e destinati al cervelletto. Gli stimoli raccolti dai tessuti periferici sono trasmessi ai nuclei cerebellari e alla corteccia cerebellare dello spinocerebellum da quattro diversi fasci nervosi a seconda delle regioni di provenienza. A livello spinale sono presenti i fasci spino-cerebellare dorsale, rostrale e ventrale, che decorrono nella sostanza bianca del cordone laterale. Ad essi si aggiunge il fascio cuneo-cerebellare che decorre nella parte dorsale del tegmento del midollo allungato. I fasci spino-cerebellare dorsale e ventrale veicolano informazioni provenienti dai meccanocettori che si distribuiscono agli arti posteriori. I fasci cuneo-cerebellare e spino-cerebellare rostrale trasmettono informazioni da recettori i cui prolungamenti periferici si distribuiscono agli arti anteriori e alle regioni del collo. Gli assoni spino-cerebellari terminano a livello della corteccia cerebellare e dei nuclei cerebellari interpositi. Essi raggiungono il cervelletto attraverso i peduncoli cerebellari rostrali (fascio spino-cerebellare ventrale) o caudali (i tre rimanenti). Nella corteccia cerebellare gli assoni spino-cerebellari danno origine a una categoria di fibre muscoidi (fibre muscoidi spino-cerebellari) nello strato dei granuli.
I fasci spino-talamici costituiscono una delle più importanti vie esterocettive coscienti. Sono spesso trattati insieme fra loro come un unico fascio spino-talamico. Come per i fasci spino-cerebellari, i neuroni di origine sono neuroni di secondo ordine presenti nella sostanza grigia delle corna dorsali del midollo spinale (lamine I, III, IV e V). Si tratta di neuroni di proiezione che, oltre alle sinapsi provenienti dai neuroni di primo ordine meccanocettivi, termocettivi e nocicettivi di piccole dimensioni (tipo B) situati nei gangli spinali, ricevono anche sinapsi eccitatorie e/o inibitorie principalmente dagli interneuroni situati nella lamina II (sostanza gelatinosa) della sostanza grigia. Questi interneuroni esercitano un’importante funzione di controllo “a cancello” sulla trasmissione sensitiva. Tali cellule, infatti, determinano una soglia dinamica di “sbarramento” per gli stimoli sensitivi prima che questi possano raggiungere il tronco cerebrale e, di qui, essere veicolati al talamo. Attraverso questo meccanismo è quindi regolato il flusso di informazioni nocicettive destinato a raggiungere la corteccia cerebrale. Gli assoni dei neuroni di origine dei fasci spino-talamici si incrociano attraversando la linea mediana a livello della commessura grigia anteriore e formano il fascio spino-talamico ventrale e laterale che si riuniscono fra loro e decorrono nella sostanza bianca del cordone laterale per poi portarsi nel tronco cerebrale e costituire il lemnisco spinale. Gli assoni del fascio spino-talamico ventrale trasmettono informazioni tattili non discriminative (tatto grossolano) e pressorie, quelli del fascio spino-talamico ventrale informazioni termiche e dolorifiche. Entrambi terminano formando sinapsi con i neuroni di terzo ordine del nucleo ventrale postero-laterale del talamo.
Il fascio spino-reticolare è la via somatosensitiva filogeneticamente più antica. Le fibre che lo costituiscono originano da neuroni di proiezione (neuroni di secondo ordine) delle lamine V e VII della sostanza grigia spinale, che ricevono sinapsi dagli stessi neuroni di primo ordine dei gangli spinali che contattano i neuroni spino-talamici. Le fibre spino-reticolari, quindi, abbandonano la sostanza grigia per portarsi nella parte inferiore del funicolo laterale. Raggiunto il tronco cerebrale, contraggono sinapsi, non incrociandosi, con i neuroni della sostanza reticolare (neuroni di III ordine) senza presentare una disposizione somatotopica. Il fascio conduce stimoli sensitivi responsabili dell’attivazione della corteccia cerebrale nei momenti di eccitazione, in alcuni stati emozionali e nel passaggio sonno-veglia in risposta al dolore o alla temperatura.
Il fascio spino-olivare è costituito dagli assoni di alcuni neuroni di proiezione situati nella porzione mediale del nucleo proprio della sostanza grigia spinale (neuroni di secondo ordine), che, dopo essersi incrociati, passano in posizione superficiale nella parte ventrale del funicolo laterale dove formano. Raggiunto il midollo allungato, questi assoni terminano formando sinapsi con i neuroni di secondo ordine del nucleo olivare inferiore. Da quest’ultimo originano le fibre olivo-cerebellari, che raggiungono la corteccia cerebellare sotto forma di fibre rampicanti. I neuroni del fascio spino-olivare ricevono afferenze tattili dai neuroni di primo ordine dei gangli spinali e le trasmettono al nucleo olivare inferiore. Queste afferenze, sperimentalmente verificate in diversi mammiferi, sono ritenute importanti perché consentono al cervelletto di intervenire sulla coordinazione muscolare quando, nel corso di un movimento, si incontrano ostacoli improvvisi.
Il fascio spino-tettale, situato in posizione profonda nella parte ventrale del funicolo laterale, è formato dagli assoni di alcuni neuroni di proiezione delle lamine III-V e VII-VIII della sostanza grigia spinale (neuroni di secondo ordine). Raggiunge il collicolo anteriore e, qui, gli assoni che lo compongono contraggono sinapsi con i neuroni del corpo genicolato laterale (neuroni di terzo ordine). Trasmette informazioni provenienti dai meccanocettori somatici (neuroni di primo ordine dei gangli spinali) che si integrano con informazioni provenienti dalle vie ottiche. Interviene nella regolazione dei movimenti di orientamento spaziale della testa.

Se vuoi saperne di più e capire meglio scorri la pagina verso il basso
I fasci spino-cerebellari e le atassie ereditarie nel cane
Le atassie ereditarie nel cane sono un ampio gruppo di malattie caratterizzate da una disfunzione spino-cerebellare o cerebellare. Sebbene ogni singola malattia sia rara, nell’insieme sono una causa importante di disturbi del movimento nei cani di razza.
Nell’uomo, la classificazione delle atassie ereditaria si basa oggi su criteri di identificazione genetica. Precedentemente la classificazione era di tipo anatomo-patologico e questo è il criterio che ancora oggi prevale in medicina veterinaria. Utilizzando tale criterio si distinguono atassie con degenerazione corticale cerebellare, degenerazione spino-cerebellare, degenerazione multipla, atassie cerebellari senza neurodegenerazione significativa e atassie episodiche. I test genetici per queste malattie sono in via di sviluppo e contribuiranno a ridurre la prevalenza della malattia, giungere a una diagnosi definitiva e a identificare potenziali terapie.
Le atassie da degenerazione corticale cerebellare (note anche come abiotrofie cerebellari, o semplicemente atassie cerebellari) sono caratterizzate dal fatto che la neurodegenerazione è in gran parte limitata alla corteccia cerebellare. Il termine abiotrofia cerebellare dovrebbe essere evitato perché “abiotrofia” significa carenza di sostanza nutritiva, ma è ormai noto che molte di queste malattie possono derivare da anomalie non metaboliche in origine. Questi disturbi possono essere ulteriormente suddivisi fra quelli che colpiscono principalmente i neuroni del Purkinje o i granuli cerebellari (degenerazione granulopriva).
Nelle atassie da degenerazione spino-cerebellare è presente un coinvolgimento del midollo allungato e/o del midollo spinale con o senza interessamento del cervelletto.
Nelle atassie da degenerazione multipla, il coinvolgimento riguarda anche i nuclei olivari, la sostanza nera e il putamen-caudato.
Nelle atassie cerebellari senza neurodegenerazione sono presenti segni clinici evidenti di disfunzione cerebellare in assenza di lesioni istopatologiche evidenti. Nelle atassie episodiche si verificano episodi di atassia profonda, sempre senza alterazioni istopatologiche. Gli individui affetti sono neurologicamente normali negli intervalli di tempo tra i diversi episodi.
| Razza | Età di insorgenza | Progressione | Gene/Locus | Modalità di trasmissione | Test genetico |
|---|---|---|---|---|---|
| Parson Russel Terrier | 6-12 mesi | Variabile | CAPN1 | Eredità poligenica | SI |
| Jack Russel Terrier | 2-12 mesi | Variabile | KCNJ10 | Eredità poligenica | SI |
| Fox Terrier a pelo liscio | 4-6 mesi | Rapida e lenta | ? | Autosomica recessiva | NO |
| Bretone spagnolo | 5-11 anni | Lenta | ? | – | NO |
La degenerazione spino-cerebellare è stata segnalata in una serie di razze canine (vedi Tabella) che costituiscono il cosiddetto gruppo dei Russell Terrier. I cani affetti sviluppano in modo drammatico deficit neurologici che iniziano con una lieve spasticità sottile dell’arto toracico e ipermetria per progredire per un periodo di mesi fino ai classici segni cerebellari che interessano tutti e quattro gli arti con atassia del tronco e tremori intenzionali. Con l’ulteriore progressione, si osserva estensione progressiva del collo e compare un’andatura “di saluto” negli arti toracici e, alla fine, i cani si muovono con il collo esteso per tenere la testa ben aderente al suolo e gli arti toracici vengono sollevati sopra la testa ad ogni passo. Le lesioni sono limitate al cervelletto, al midollo allungato e al midollo spinale.
C’è una massiccia perdita di neuroni del Purkinje, di neuroni dei nuclei cerebellari profondi accompagnata da degenerazione neuronale nei nuclei gracile e cuneato e degenerazione assonale nelle colonne dorsali e nelle aree laterali e ventromediali del cordone laterale del midollo spinale.
La causa genetica e la modalità di trasmissione non sono state identificate.
Il gruppo dei Terrier si presenta con una gamma di segni molto diversificata e solo di recente, con l’identificazione di due diverse mutazioni, è diventato chiaro che ci sono almeno due diverse malattie che causano atassia ereditaria in queste razze. Il gruppo Russell Terrier include Jack Russell, Parson Russell e Russell Terrier; si ritiene che tutti discendano da cani di proprietà di un solo allevatore.
Il primo fenotipo importante è un’atassia spino-cerebellare (SCA) che insorge tra i 2 e i 10 mesi di vita combinata con miochimia (contrazioni muscolari involontarie increspate), neuromiotonia (contrazioni muscolari sostenute) e convulsioni. La sindrome è ora identificata utilizzando il termine SAMS (Spinocerebellar Ataxia with Myokymia, Seizures or both = atassia spinocerebellare con miochimia, convulsioni o entrambi). In genere c’è una progressione evidente dei segni clinici nell’arco di alcune settimane, poi una stabilizzazione con periodi intermittenti di deterioramento clinico. I cani mostrano un’andatura ipermetrica, spastica, con reazione posturale e deficit del posizionamento propriocettivo localizzati al midollo spinale cervicale. La maggior parte dei cani viene soppressa a causa della malattia in media un anno dopo l’esordio, sebbene siano riconosciuti anche fenotipi più lievi, più lentamente progressivi.
I reperti patologici di un’assonopatia sono più gravi nel midollo spinale, ma sono presenti in tutto l’encefalo. Si osservano gliosi, rigonfiamento degli assoni e perdita di fibre mieliniche nei tratti spino-cerebellari, nel lemnisco laterale. Inoltre esiste una degenerazione dei nuclei cocleari e del corpo trapezoide.
L’esistenza di diverse espressioni fenotipiche della stessa malattia è stata
confermata dall’identificazione di una mutazione nel gene KCNJ10 nei soggetti colpiti. Il gene KCNJ10 codifica per il canale del potassio (K+) rettificatore entrante Kir4.1 che è espresso dalle cellule gliali. Kir4.1 è responsabile del potenziale di membrana a riposo negativo degli astrociti e della grande conduttanza per K+ attraverso le membrane degli astrociti. Queste proprietà consentono agli astrociti di tamponare le fluttuazioni del K+ extracellulare causate dalla depolarizzazione assonale, e la disfunzione di questi canali provoca un aumento del K+ extracellulare con conseguente ipereccitabilità. Anche i trasportatori del glutamato si affidano a questa elevata conduttanza per K+ per rimuovere il glutammato dalle sinapsi, quindi la disfunzione di Kir4.1 può anche indurre ipereccitabilità attraverso la mancata rimozione del glutamato a livello delle sinapsi. Per di più, Kir4.1 è importante anche nel mantenimento dell’integrità della mielina nel SNC, sebbene il meccanismo alla base di tale effetto sia al momento non chiaro. La SAMS può essere confermata con test genetici (vedi Tabella).
La seconda atassia ereditaria che si osserva nei Parson Russell Terrier provoca una SCA pura con un’età di esordio compresa tra 6 e 12 mesi, denominata atassia ad insorgenza tardiva nelle pubblicazioni più vecchie. Questa malattia è stata associata a una mutazione missenso in CAPN1. Il gene CAPN1 codifica per una cisteina proteasi intracellulare calcio-dipendente chiamata calpaina 1. Sebbene l’esatta funzione della calpaina 1 sia sconosciuta, si suppone essa svolga un ruolo nel mantenimento e nel rimodellamento neuronale. Questo può provocare un terzo disturbo in cui potenziali difetti nello smaltimento delle proteine a livello della cellula nervosa ne provoca la degenerazione. Anche la SCA pura può essere diagnosticata con un test genetico.
Bibliografia: Urkasemsin G, Olby N (2014) Canine hereditary ataxia. Vet Clin Small Anim 44:1075–1089
Prova a rispondere a queste domande:
1. I cani colpiti da SCA possono presentare potenziali evocati uditivi anomali al BAER Test (audiometria a risposta evocata del tronco encefalico). Il BAER test si esegue registrando con elettrodi posti sulla volta cranica le risposte ad uno stimolo sonoro. Perché i cani atassici possono presentare risposte anomale al test?
2. Cos’è il lemnisco laterale?
3. Perché nella SCA le lesioni della sostanza bianca spinale si localizzano nelle parti superficiali del cordone laterale?
4. Come terminano i fasci spino-cerebellari a livello del cervelletto?
5. Perché i cani affetti da SCA presentano deficit del posizionamento propriocettivo?
(le risposte sono al fondo della pagina)

Ricordati di consultare un libro o cercare informazioni in rete se non conosci il significato di alcuni termini!
Risposte
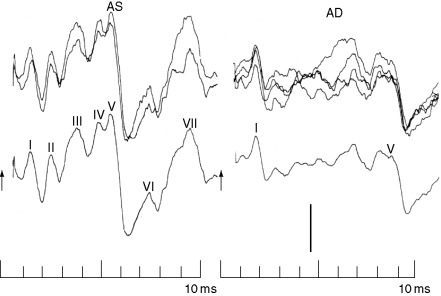
1. Le alterazioni alle risposte ai potenziali evocati nel BAER test dipendono dalla possibile degenerazione dei nuclei cocleari e del corpo trapezoide, che fanno parte delle vie acustiche centrali. I potenziali evocati uditivi del tronco cerebrale (BAEP) sono una serie di onde registrate entro 10 ms da uno stimolo uditivo che vengono generate all’interno delle strutture che costituiscono le vie acustiche centrali del tronco cerebrale. Lo stimolo preferito è un clic a banda larga con un livello di pressione sonora di picco di 90–120 dB, sebbene possano essere utilizzati toni puri o altri segnali acustici. Le forme d’onda utilizzate clinicamente sono chiamate onde I, III e V. L’elettrococleogramma si verifica prima dell’onda I. Si pensa che l’onda I sia generata nella parte distale del n. acustico (VIII), l’onda III nel ponte all’interno o vicino al nucleo olivare craniale e l’onda V nella parte aborale del mesencefalo vicino al collicolo caudale. I criteri principali per l’anomalia clinica sono i ritardi nelle onde I-V o altre latenze interonda, differenze eccessive di latenza tra le onde e il rapporto di ampiezza fra onda V e onda I.
2. Il lemnisco laterale è un tratto di assoni situato nel tronco cerebrale che trasporta informazioni sonore dai nuclei cocleari a vari nuclei del tronco cerebrale e infine al collicolo caudale controlaterale del mesencefalo. Intercalati all’interno di queste fibre si trovano tre gruppi distinti di neuroni, principalmente inibitori, chiamati nuclei del lemnisco laterale.
3. Perché le lesioni della sostanza bianca spinale si localizzano in corrispondenza della posizione dei fasci spino-cerebellari (vedi figura a inizio pagina).
4. I fasci spino-cerebellari raggiungono il cervelletto attraverso i peduncoli cerebellari e terminano nella corteccia cerebellare costituendo le fibre muscoidi spino-cerebellari.
5. I cani affetti da SCA presentano un deficit propriocettivo perché la degenerazione assonale e neuronale può colpire, rispettivamente, i cordoni dorsali della sostanza bianca e i neuroni dei nuclei gracile e cuneato. Ne consegue un malfunzionamento della via della colonna dorsale/lemnisco mediale che veicola stimoli propriocettivi provenienti dai muscoli e dalle articolazioni del collo, del tronco e degli arti.
